|
Sticklebacks offer a powerful new system for studying the molecular basis
of evolutionary change in many different natural populations
(Kingsley and Peichel 2007).
These fish have undergone one of the most recent and dramatic evolutionary
radiations on earth, colonizing countless new freshwater lake and stream
environments generated at the end of the last Ice Age approximately 10,000
years ago. Many recently evolved freshwater populations show dramatic
differences in morphology, physiology, and behavior. Although often
reproductively isolated in nature, different stickleback species can still be
crossed using artificial fertilization in the laboratory. This provides a
unique opportunity to identify the key chromosome regions, genes, and mutations
that control repeated evolution of new traits and new species under a full
range of fitness constraints in the wild.
Thousands of papers and several full-length textbooks have been written on
the ecology, morphology, paleontology, and adaptive significance of stickleback
traits. We have developed a complete set of genetic and genomic resources for
this classic system, including the first genome-wide linkage maps, transgenic
methods, expressed sequence tag (EST) collections, large-insert BAC (bacterial
artificial chromosome) libraries, and physical maps useful for positional
cloning (genetic work done in collaboration with Dolph Schluter [University of
British Columbia–Vancouver]; molecular work done in collaboration with
Jane Grimwood, Jeremy Schmutz, and Richard Myers [Hudson Alpha Institute];
Chris Amemiya [Benaroya Research Institute at Virginia Mason, Seattle];
Pieter de Jong [BACPAC Resources, Oakland, California]; and Marco Marra and
Jacqueline Schein [University of British Columbia]). We also nominated
threespine sticklebacks for complete genome sequencing to the National
Institute of Human Genome Research (Kingsley 2003).
We have worked with the Broad Institute and Ensembl to develop, assemble, and
annotate the first whole-genome sequence assembly for Gasterosteus aculeatus,
and to resequence additional stickleback genomes from many interesting
populations around the world
(Jones et al. 2012).
The high-quality reference genome can be browsed using the UC-Santa Cruz and
ENSEMBL genome browsers, and comparative results based on re-sequencing 21
other stickleback genomes can also be explored and downloaded at
"sticklebrowser"
(Jones et al. 2012).
We are using these new tools to identify the number, location, and type of
genes and mutations that control major evolutionary differences in body size
and color, skeletal armor, feeding modifications, fin development, behavioral
characteristics, and physiological traits such as temperature preference and
salinity tolerance. Our studies have focused on a number of populations that
have been particularly well studied from a morphological and ecological
perspective, including fish from lakes near Vancouver (in collaboration with
Dolph Schluter, University of British Columbia, Vancouver); in Alaska
(with Michael Bell, Stony Brook); the Haida Gwaii Islands (with Thomas
Reimchen, University of British Columbia–Victoria); Iceland (with
Bjarni Jónsson, Institute of Freshwater Fisheries, Iceland); and other
populations in California, Washington State, Nova Scotia, and Scotland.
Charles Darwin and later population geneticists have predicted that most
interesting evolutionary adaptations in nature will be controlled by large
number of genetic changes with infinitesimally small effects. In contrast,
our linkage studies have shown that large evolutionary changes in sticklebacks
often map to some chromosomes with surprisingly large effects
(Colosimo et al. 2004;
Shapiro et al. 2004;
Miller et al. 2007;
Albert et al. 2008;
Kitano et al. 2009;
Chan et al. 2010;
Greenwood et al. 2011;
Wark et al. 2012;
Cleves et al. 2014;
Miller et al. 2014;
Indjeian et al. 2016).
Using positional cloning methods, we have now identified the genes responsible
for some of the dramatic morphological changes between populations. For
example, loss of the entire pelvic apparatus in some populations is controlled
by changes in a master regulatory homeodomain transcription factor (Pitx1) that
is normally expressed in hindlimbs but not forelimbs of most vertebrates (the
homeodomain protein Pitx1; Shapiro et al. 2004;
Chan et al. 2010).
Similarly, large differences in armor plate patterning are controlled by
changes in the developmental signaling gene Ectodysplasin (EDA)
(Colosimo et al. 2005);
and changes in body pigmentation are controlled by changes in the major stem
cell factor signaling gene Kitlg (Miller et al. 2007).
In all three of these cases, null mutations of the corresponding genes in mice
or humans cause major developmental defects or lethality. However, evolution
has been able to use these genes to induce major morphological changes in wild
animals, using regulatory changes rather than coding region mutations to
confine dramatic differences to particular body regions
(Shapiro et al. 2004;
Miller et al. 2007;
Chan et al. 2010;
O'Brown et al. 2015).
Our work on stickleback evolution began with genome-wide linkage mapping,
followed by detailed case studies of a handful of major genes controlling
large morphological changes. Using the lessons learned from these initial
examples, we have recently been able to identify a large genome-wide set of
loci that are repeatedly selected when marine fish colonize and adapt to new
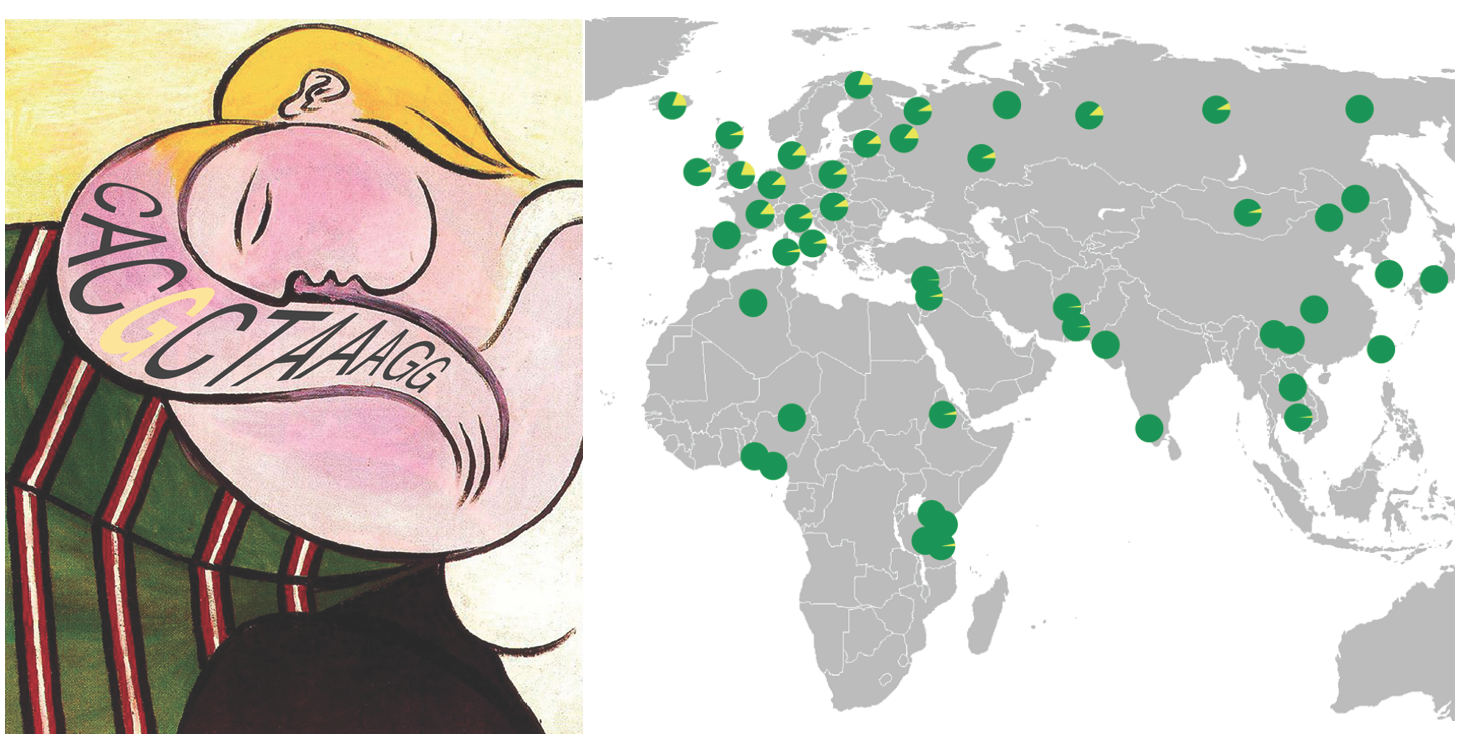
environments (Jones et al. 2012).
With hundreds of adaptive loci now in hand from genome-wide re-sequencing
studies we can now begin to address quite general questions about the genomic
mechanisms contributing to evolutionary change (Jones et al. 2012).
Our genome-wide studies show that chromosome inversions can play an important
role in maintaining suites of differences between marine and freshwater fish.
And, based on a large number of examples, we find that both protein-coding
(Marques et al. 2017) and
regulatory changes contribute to adaptive evolution, with regulatory changes
predominating by about 4 to 1 during evolution of natural stickleback populations
(Jones et al. 2012).
Finally, the widespread evolution of sticklebacks offers a unique
opportunity to test whether the same or different genes are used when the same
traits evolve in widely separated locations. Genetic mapping, complementation
tests, gene expression studies, and genome-wide re-sequencing studies all show
that similar genetic mechanisms are used when similar traits are selected in
multiple populations around the world (Colosimo et al. 2005;
Shapiro et al. 2006;
Miller et al. 2007;
Chan et al. 2010;
Jones et al. 2012;
O'Brown et al. 2015;
Indjeian et al. 2016;
Marques et al. 2017).
How far might such reuse of particular genes extend? Our recent studies
suggest that the genes underlying major morphological change in sticklebacks
are also reused when similar morphological changes evolve even in distantly
related animals, including loss of hindlimbs in marine mammals
(Shapiro et al. 2006),
changes in bone size in humans (Indjeian et al. 2016),
and recent changes in skin and hair color in humans that have colonized new
environments around the world (Miller et al. 2007;
Guenther et al. 2014).
We also find that the same type of regulatory changes that appear particularly
important during stickleback evolution have also contributed to both loss and
gain traits in the human lineage (McLean et al. 2011;
Capellini et al. 2017).
Further studies of sticklebacks may thus reveal general features of
evolutionary change, with broad implications for our understanding of evolution
in many other vertebrates, including humans.
More information on research projects in mice, human evolution, and human disease.
|