|
At early stages of our research, we helped elucidate simple
Mendelian diseases in humans that affected carbohydrate metabolism and mineral
deposition in the skeleton
(Kingsley et al., 1986; Ho et al., 2000; Pendleton et al., 2002; Gurley et al., 2006).
Identifying the molecular basis of these traits was very satisfying, but protein coding
mutations in the genes were deleterious, and the corresponding alleles were relatively
rare in populations. With our recent progress on evolutionary studies, we have now
begun identifying the molecular basis of traits that had been positively selected
during human history
(Miller et al., 2007; Guenther et al., 2014; Capellini et al., 2017; Song et al., 2018).
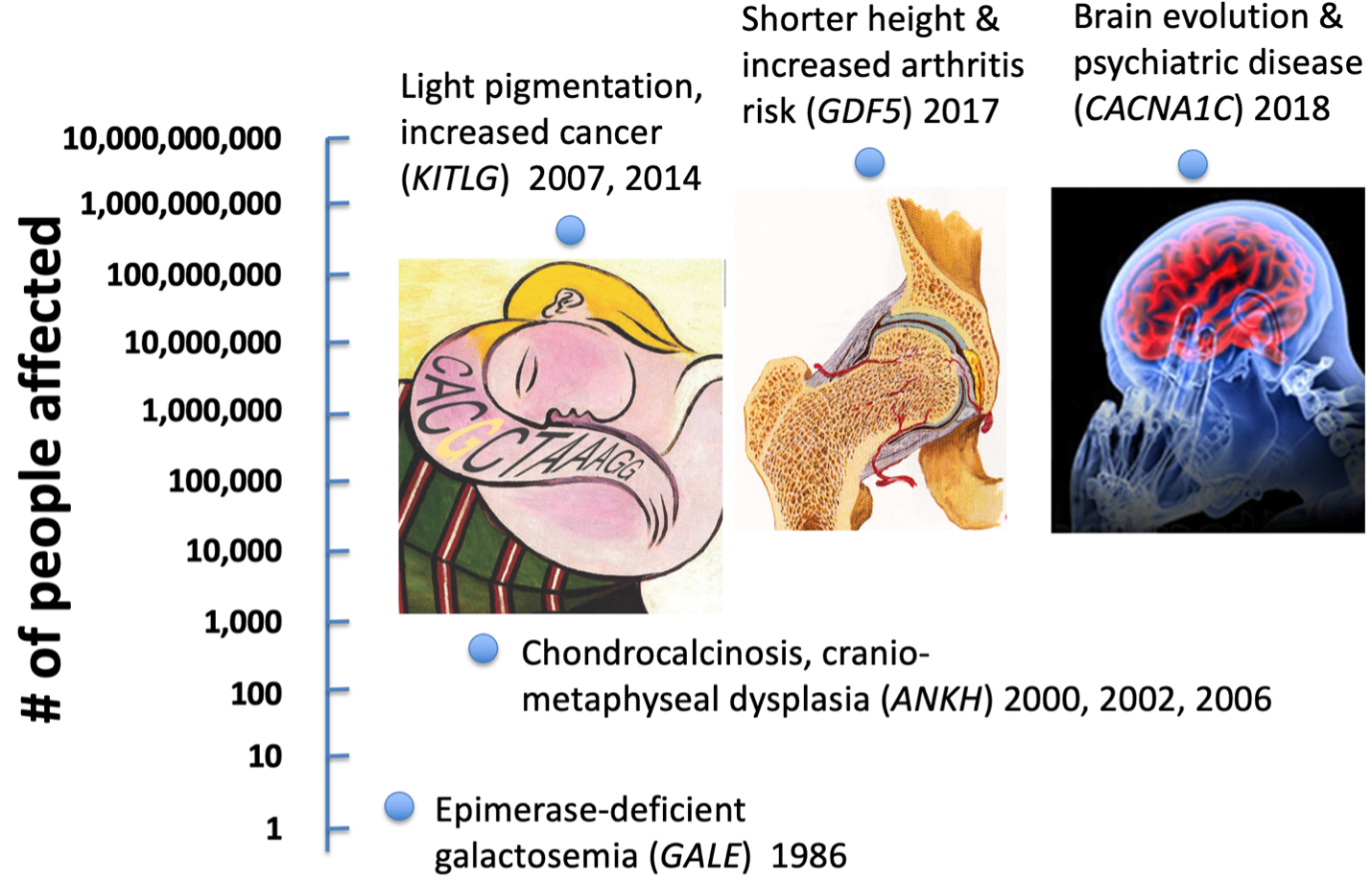
As expected, human alleles that have been positively selected are present in hundreds
of millions to billions of people around the world. Interestingly, the variant alleles
selected because they confer an advantage on particular traits often change the
susceptibility to other human diseases as well. For example, selection for light
pigmentation at northern latitudes may help maintain vitamin D production in limited
sunlight, but also increases risk of skin cancer, melanoma, or other cancers
(Miller et al., 2007; Zeron-Medina et al., 2013).
Similarly, positive selection for altered human body proportions in different environments
may increase risk of arthritis or bone fractures later in life (Capellini et al., 2017).
And rapid evolution of brain genes in the human lineage also appears to increase risk
of psychiatric diseases like schizophrenia or bipolar disease
(Srinivasan et al., 2016; Song et al., 2018). Thus the search for the molecular basis
of human evolutionary traits has surprising relevance some of the most common medical
disorders in modern populations. Positively selected alleles become extremely common
alleles, and the specific regulatory variants of KITLG, GDF5, and CACNA1C we are
characterizing are now among the world's most important population risk factors for
human cancer, arthritis, and psychiatric diseases.
Capellini, T.D., Chen, H., Cao, J., Doxey, A.C., Kiapour, A.M., Schoor, M., and Kingsley, D.M. (2017). Ancient selection for derived alleles at a GDF5 enhancer influencing human growth and osteoarthritis risk. Nat. Genet. 49, 1202-1210.
Guenther, C.A., Tasic, B., Luo, L., Bedell, M.A., and Kingsley, D.M. (2014). A molecular basis for classic blond hair color in Europeans. Nat. Genet. 46, 748-752.
Gurley, K.A., Reimer, R.J., and Kingsley, D.M. (2006). Biochemical and genetic analysis of ANK in arthritis and bone disease. Am. J. Hum. Genet. 79, 1017-1029.
Ho, A.M., Johnson, M.D., and Kingsley, D.M. (2000). Role of the mouse ank gene in control of tissue calcification and arthritis. Science 289, 265-270.
Kingsley, D.M., Krieger, M., and Holton, J.B. (1986). Structure and function of low-density-lipoprotein receptors in epimerase-deficient galactosemia. N. Engl. J. Med. 314, 1257-1258.
Miller, C.T., Beleza, S., Pollen, A.A., Schluter, D., Kittles, R.A., Shriver, M.D., and Kingsley, D.M. (2007). cis-Regulatory changes in Kit ligand expression and parallel evolution of pigmentation in sticklebacks and humans. Cell 131, 1179-1189.
Pendleton, A., Johnson, M.D., Hughes, A., Gurley, K.A., Ho, A.M., Doherty, M., Dixey, J., Gillet, P., Loeuille, D., McGrath, R., et al. (2002). Mutations in ANKH Cause Chondrocalcinosis. Am. J. Hum. Genet. 71, 933-940.
Song, J.H.T., Lowe, C.B., and Kingsley, D.M. (2018). Characterization of a human-specific tandem repeat associated with bipolar disorder and schizophrenia. Am. J. Hum. Genet. 103, 421-430.
Srinivasan, S., Bettella, F., Mattingsdal, M., Wang, Y., Witoelar, A., Schork, A.J., Thompson, W.K., Zuber, V., Winsvold, B.S., Zwart, J.-A., et al. (2016). Genetic markers of human evolution are enriched in schizophrenia. Biol. Psychiatry 80, 284-292.
Zeron-Medina, J., Wang, X., Repapi, E., Campbell, M.R., Su, D., Castro-Giner, F., Davies, B., Peterse, E.F.P., Sacilotto, N., Walker, G.J., et al. (2013). A polymorphic p53 response element in KIT Ligand influences cancer risk and has undergone natural selection. Cell 155, 410-422.
More information on research projects in mice, sticklebacks, and human evolution. |